מחלות מיאלופרוליפרטיביות (MPD - (Myeloproliferative disorders - הן קבוצת מחלות ממאירות לא אחידות, המאופיינת על ידי שגשוג תאים משורה אריטרוידית, מגקריוציטית או מיאלוידית. קבוצה זו כוללת שלוש מחלות קלאסיות: פוליציטמיה ורה (Vera-PV (Polycythemia, טרומבוציטוזיס ראשונית, ET (Essential Thrombocytosis) מיאלופיברוזיס (MF (Myelofibrosis ומספר מחלות נדירות נוספות. לויקמיה מיאלוידית כרונית (CML) שויכה בעבר ל-MPD, אך כיום היא נחשבת למחלה נפרדת המאופיינת על ידי כרומוזום פילדלפיה (Ph) ו/או טרנסלוקציה BCR/ABL. מחלות מיאלופרוליפרטיביות שאינן CML נקראות Ph(-) MPD. מחלות אלו הן מחלות המטואונקולוגיות חד שבטיות של תאי גזע המטופואטיים.
מחלות מיאלופרוליפרטיביות
פוליציטמיה ורה - חולי PV מתלוננים לעתים על כאבי ראש, סחרחורת וגרד (בעיקר לאחר מגע עם מים חמים) אך רוב החולים הם א-תסמיניים. בבדיקה גופנית ניתן למצוא סומק בפנים ולעתים טחול מוגדל. בבדיקות מעבדה נמצאים ערכים גבוהים של אריטרוציטים (RBC), המוגלובין (HB), המטוקריט (HCT), ליקוציטים (WBC), טסיות דם (PLT, LDH), חומצת שתן וויטמין B12. רמת האריטרופואטין (EPO) בדם היא נמוכה ברוב המקרים. הסיבוכים השכיחים ב-PV הם נטייה לדימומים ולטרומבוזות עורקיות ורידיות וקיים סיכון נדיר להתמרה ל-MF או ללוקמיה חריפה.
הטיפול ב-PV כולל הקזות דם ו/או טיפול כימותרפי על ידי Hydroxyurea. מטרת הטיפול ב-PV היא להוריד את רמת ה-HCT מתחת ל-45 אחוז אצל גברים ולפחות מ-42 אחוז אצל נשים. טיפול בהקזות מומלץ בצעירים עקב חשש לסיכון לויקמוגני של הטיפול ב-Hydroxyurea. בשנים האחרונות הוכח שטיפול באספירין במינון נמוך מפחית את הסיכון לטרומבוזות ללא עלייה משמעותית בשיעור הדימומים.
טרומבוציטוזיס ראשונית - ET היא מחלה המאופיינת על ידי עלייה במספר הטסיות ללא שנמצאה סיבה אחרת לטרומבוציטוזיס. חולי ET עלולים לסבול הן מטרומבוזות והן מדימומים. אירועים טרומבוטיים שכיחים יותר כאשר מספר הטסיות עולה על dl1,000x103. דימומים לעומת זאת שכיחים יותר כאשר מספר הטסיות עולה מעל ל-1,500x103dl. ההתוויות לטיפול להורדת טסיות הן גיל מעל 60, מספר טסיות מעל 1,000x103dl או עבר של אירועים טרומבוטיים או דימומים. הטיפולים הכלליים הם: ANAGRELIN PO,
PO HYDROXYUREA או INTERFERON SC.
מיאלופיברוזיס - MF היא מחלה המאופיינת בהחלפת רקמת מוח בסיבי קולגן, המטופואזיס מחוץ למח עצם, הגדלת טחול ותמונה אופיינית במשטח דם: אריטרוציטים בצורת דמעה, הופעת אריטרובלסטים וסטייה שמאלה של השורה המיאלוידית, טרומבוציטוזיס או טרומבוציטסניה. האבחנה נעשת על ידי ביופסיית מח עצם אולם טרם נמצא טיפול יעיל למחלה זו. השתלת מח עצם אלוגנאית יכולה אולי להאריך את החיים אצל מקצת מהחולים צעירים. טיפול ב-Thalidomide במינון נמוך יכול לשפר את האנמיה של חלק מחולי MF.
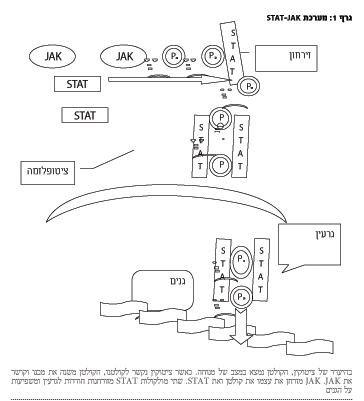
מערכת STAT-JAK ותפקידה בהמטופואזיס
התאים המטופואטיים עוברים תהליכים רבים במח עצם: התחלקות, התמיינות, הבשלה, ייצור וחילוף חומרים ומוות (אפופטוזיס). תהליכים אלה תלויים בהפעלה ובשיתוק גנים ספציפיים. פעילות הגנים מושפעת על ידי חומרים שונים כגון ציטוקינים, הורמונים, גורמי צמיחה, אינטרפרונים וכדומה. חומרים אלה נקשרים לקולטניהם על פני קרום התא. על מנת להעביר אות מקולטן לגרעין קיימת מערכת שלמה של חלבונים בציטופלזם, מערכת ה-STAT-JAK Janus Kinase ו-Signal Transducers and Activation of Transcription (מעביר אות והפעלת שעתוק).
JAK נקראת על שמו של אל רומי Janus שהיה בעל שני פרצופים. כך גם אצל החלבונים ממשפחת JAK, יש מבנה שכולל שני קטעים זהים וצמודים זה אל זה, כמו שני פרצופי האל. כיום ידוע על ארבעה מינים של משפחת JAK ביונקים: JAK1, JAK2, JAK3 ו-2TYK. JAK הם אנזימים עם פעילות של זרחון חומצה אמינית טירוזין (TK (Tyrosine Kinase. בין TK מבנה של JAK בלעדי - רק אחד מהקטעים ה"דו פרצופיים" מבטא פעולה קטליטית. לשני אין פעילות קטליטית אך הוא חיוני לוויסות פעולה קטליטית של הדומיין הראשון.
כיום ידוע על שבעה גנים שמקודדים STAT שונים. מדובר במשפחה של חלבונים ציטופלזמתיים ללא פעילות אנזימטית אשר חודרים אחרי הפעלתם לתוך גרעין. התהליך מתחיל כאשר ציטוקין נקשר לקולטנו וגורם לדימריזציה של הקולטן. JAK נקשר לחלק ציטופלזמטי של הקולטן, עובר זרחון עצמו והפעלת עצמו ומזרחן את ה-STAT ובכך גורם להפעלתו. שני ה-STAT המופעלים נקשרים זה לזה ואז הם מסוגלים לחדור לגרעין ולהפעיל או לדכא גנים שונים, כולל אלה שחשובים להמטופואזיס תקינה.
המידע לגבי תפקידם של ה-JAK השונים בהמטופואזיס תקינה התקבל מניסויים בעכברים עם שיתוק גנים מסוימים (mice knockout).
JAK1 דרוש לאיתות מקולטנים של אינטרפרוני אלפא, בטא וגמא ואינטרליקינים שונים. עכברים עם חוסר ב-(JAK1 (mice knockout נולדים עם פגם במערכות לימפוידית ועצבית, והם קטנים ומתים בתוך ימים ספורים.
JAK2 נקשר לקולטני GM-CSF, אריטרופואטין (EPO), טרומבופואטין (TPO) ו-IL3,5. עכברי knockout ל-JAK2 מתים בתוך 12 ימים תוך רחמיים עקב כישלון האריטרופויזיס.
JAK3 הוא חיוני להעברת אות מקולטני אינטרלויקינים רבים. חיות knockout ל-JAK3 נולדות עם חוסר חיסוני עקב ירידה קשה במספר הלימפוציטים מסוג B ו-T ומחוסר ב"רוצחים טבעיים" (NK).
אחד המצבים של חוסר אימוני מולד אצל בני אנוש, חוסר אימוני קשה משולב (SCID) אוטוזומלי רצסיבי, נגרם על ידי מוטציה ב-JAK3.
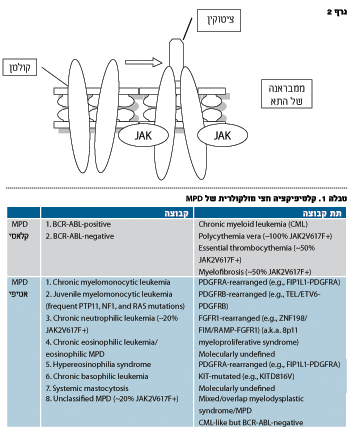
מוטציה JAK-2 V617F בפתוגנזה
של מחלות מיאלופרוליפרטיביות
חוסר תורשתי של איתות STAT-JAK מלווה בחוסר חיסוני. מוטציות נרכשות שגורמות להפעלת יתר של מערכת ה-STAT-JAK משתתפות בהתפתחות ממאירויות המטולוגיות. בשלוש השנים האחרונות התגלתה מוטציה חדשה: החלפת הגואנין לטימידין בנוקלאוטיד 1849 באקסון 14 של JAK2, גן הנמצא בזרוע הקצרה של כרומוזום 9. מוטציה זו גורמת להחלפה של חומצה אמינית לפנילאלנין בעמדת 617 ומצוינת כ-617FV2-JAK. נמצא שמוטצייה זו קיימת בקרב כ-95 אחוז מקרב חולי PV ובכמחצית מחולי ET ו-MF. לעומת זאת, מוטציה זאת נדירה בקרב החולים הסובלים מ-MPD אחרות ומתסמונת מיאלודיספלסטית.
מוטצית 617FV2-JAK ממוקדת בקטע שאינו קטליטי ("דומיין קטליטי מדומה") של חלבון JAK2. "דומיין קטליטי מדומה" ידוע כגורם המעכב את פעולת הזרחון הטירוזין של דומיין קטליטי. JAK2 ללא מוטציה מופעל רק כאשר ציטוקין (למשל EPO) נקשר עם קולטנו (EPOR). מוטצית 617FV2-JAK גורמת לכך שאנזים הופך להיות פעיל באופן קבוע או מופעל בצורה מוגזמת כתגובה לגורמים רגילים. הופעת המוטצייה על פרקורסורים אריטרוידיים מאפשרת צמיחת תאים אלה בהיעדר EPO. המנגנון של הפעלת יתר של JAK2 כולל גם תגובה מופחתת של 617FV2-JAK לדיכוי על ידי חלבוני
(Socs suppressors of cytokine signaling proteins). במצב תקין, Socs 3 מדכא את פעולת JAK2 וקולטנו של EPO. בנוכחות של 617FV2-JAK, ה-Socs 3 מגביר את הפעלת JAK2 על ידי EPO.
התפקיד הגורלי בפתוגנזה של מחלות מיאלופרוליפרטיביות הודגם בשיטה של הנדסה גנטית: עכברים פיתחו מחלה דמויית PV אחרי קרינה במנה קטלנית והשתלת מח עצם עם תאים אשר קיבלו 617FV2-JAK דרך נגיף.
JAK-2V617F במחלות מיאלופרוליפרטיביות - מבט קליני
בקרב החולים הסובלים ממחלות מיאלופרוליפטיביות, מוטציית F617V2-JAK נמצאת בתאים מיאלוידיים, בתאי אב ולפעמים גם בשורה לימפוידית. אין עדות על שוני בהתפלגות של 617FV2-JAK בשורות המטופויטיות שונות בקרב חולי PV, ET ו-MF. בשלב זה אין תשובה לשאלה מדוע מוטציה אחת אכן גורמת לשלוש מחלות שונות.
יש לציין שבקרב חולי ET עם 617FV2-JAK נמצאו רמות המוגלובין והמטוקריט מעט גבוהות וטסיות מעט נמוכות בהשוואה עם ET ללא 617FV2-JAK. עובדה זו מעוררת את השאלה האם ET בעלת המוטציה היא שלב מוקדם של פוליציטמיה ורה?
מטופלים רבים הם הטרוזיגוטיים ל-617FV2-JAK, כך שקיימת תת אוכלוסיה של תאים הומוזיהוטיים ל-617FV2-JAK מעורבת עם תת אוכלוסיה של תאים ללא מוטציה, או אוכלוסיה של תאים עם אלל חד עם 617FV2-JAK ושני ללא מוטציה. רוב חולי PV הם הומוזיהוטיים ל-617FV2-JAK, כאשר רוב חולי ET הם הטרוזיהוטיים.
האבחנה המבדלת בין מחלות מיאלופרוליפרטיביות שונות אינה פשוטה ולא תמיד קל להבדיל בין PV, ET ו-MF. בנוסף, לא תמיד קל להבדיל בין PV ואריטרוציטוזיס משנית, לבין ET לבין טרומבוציטוזיס תגובתית. כיוון שלא נמצא 617FV2-JAK בקרב חולים עם אריטרוציטוזיס משנית או טרומבוציטוזיס תגובתית, נוכחות ה-617FV2-JAK תומכת מאוד באבחנה של מחלה מיאלופרוליפרטיבית.
העדר מוטציה מסוג JAK אצל חולה אריטרוציטוזיס שוללת את האפשרות של PV באופן כמעט מוחלט, אך היעדר המוטציה אצל מטופלים עם חשד ל-ET או MF אינו שולל אפשרות של מחלות אלו. מספר מחקרים קליניים הראו הישרדות נמוכה יותר בקרב חולי MF עם 617FV2-JAK על רקע שיעור גבוה יותר של התמרה ללוקמיה חריפה, אולם, התוצאות אינן חד משמעיות לגבי הנטייה לטרומבוזות. מספר מחקרים הדגימו עלייה בשיעור טרומבוזות עורקיות ולא ורידיות בקרב חולי ET ו-MF חיוביים ל-617FV2-JAK ומחקרים אחרים לא מצאו שום הבדל בין החולים החיוביים או השליליים למוטציה.
קלסיפיקציה חדשה של מחלות מיאלופרוליפרטיביות
את המחלות המיאלוידיות הממאירות ניתן לסווג לשתי קבוצות גדולות: לוקמיה מיאלוידית חריפה (AML) ומחלות מיאלוידיות כרוניות. ה-AML מאופיינת על ידי נוכחות של יותר מ-20 אחוז בלסטים מיאלוידיים בדם או במח עצם. מחלות מיאולידיות כרוניות ניתן לחלק למחלות מיאלופרוליפרטיביות (MPD) ולמחלות מיאלודיספלסטיות (MDS).
לפני כ-55 שנה Dameshek חשב ש-PV, ET, MF ו-CML הן מחלות קרובות וקרא להן MPD. כאשר נמצא סמן מולקולרי ל-CML (טרנסלוקציה ABL-BCR), המחלה הוצאה מרשימת ה-MPD. כיום, לאור גילוי סמן מולקולרי נוסף, ה-617FV2-JAK ברוב חולי PV ולמחצית מחולי ET ו-MF, התעורר שוב הצורך במיון מחדש של המחלות. Tefferi הציעה קלסיפיקציה חדשה של MPD הכוללת MPD קלאסיות ו-MPD אטיפיות לפי ההפרעות המולקולריות הידועות (טבלה1).
פיתוח טיפול מכוון ל-JAK-2V617F
(Imatinib (Glivec הוא מעכב ראשון של אנזים מקבוצה של TK. התרופה פותחה כמעכב של ABL-BCR וגרמה למהפך בטיפול ב-.Glivec
CML משפיע גם על TK אחרים כמו PDGFR ודרך עיכובם נמצא יעיל גם בתסמונת היפראוזינופילי (HES) עם טרנסלוקציה PDGFRA-FIPIL1 ובחלק חולי עם לוקמיה מיאלומונוציטית כרונית (CML) עם מוטציית PDGFRB. Glivec והתרופות מהדור הבא כנגד CML אינם משפיעים על JAK2. לאור הממצא החשוב של המוטציה החדשה במערכת ה-JAK, אין לשלול התפתחות תרופות ספציפיות המכוונות למערכת ה-JAK כבר בעתיד הקרוב.
ד"ר א. נמץ, פרופ' ג. לוגסי, מחלקה המטולוגית ע''ש יוסף מיכאלי, מרכז רפואי ברזילי, אשקלון |