אנמיה ע"ש פנקוני (א"פ), שתוארה על ידי Guido בשנת 1927, היא מחלה תורשתית נדירה אך השכיחה ביותר בקבוצת המחלות עם כשלון מח עצם תורשתי. שכיחות המחלה נעה בין מקרה אחד לחמישה מקרים למיליון. תורשת המחלה היא אוטוזומלית רצסיבית, למרות שיש מספר קטן של חולים שמחלתם אחוזה במין. המחלה מתאפיינת בהופעת מומים מולדים, כשלון מתקדם של מח העצם וכן נטייה להופעה של גידולים ממאירים.
ב-20 שנים האחרונות חלה התקדמות בהבנת הביולוגיה של המחלה והתברר שא"פ היא מחלה הטרוגנית שנגרמת על ידי פגמים בלפחות 13 גנים (A, B, C, D1, D2, E, F, G, I, J, L, M, N). החלבונים המקודדים על ידי גנים אלה הם חלק מקבוצת חלבוני התא שתפקידם לתקן נזקים בדנ"א ולשמור על יציבות הגנום. חסרונם מביא לתמונה הקלינית הקשה של א"פ. בסקירה זו נתאר את ההתקדמות בהבנת המחלה ואת השיפור באבחון, בטיפול ובמניעה.
התפקיד התאי של חלבוני פנקוני בשמירת היציבות הגנומית
תאים מחולי א"פ מראים דרגה גבוהה של שבירות כרומוזומים עצמונית וזו עולה בצורה משמעותית עם החשיפה לחומרים שקושרים גדילי דנ"א כמו iepoxybutane) DEB). תכונה זו של חוסר יציבות גנומית הביאה כבר לפני יותר מ-20 שנה לפתח בדיקה אבחנתית למחלה (שבירות כרומוזומים בתאי חולים לפני ואחרי הוספת חומרים קושרי גדילי דנ"א כ-DEB או מיטומיצין C).
ההתקדמות הראשונית בהבנת המחלה חלה כאשר הוכנס מקטע דנ"א הכולל את הגן החסר לתרבית תאי החולה ותוקנה הרגישות לחומרים יוצרים קשרים בין גדילי הדנ"א. כך הוגדרו 13 קבוצות השלמה (complementation groups) שכל אחת מייצגת גן אחר. בעזרת צמצום גודל מקטע הדנ"א המאפשר תיקון שבירות הכרומוזומים ועל ידי תאחיזה גנטית, זוהו 13 הגנים המעורבים במחלה. הגנים שזוהו עד כה הם FANCA, FANCB, FANCC, FANCD1, FANCD2, FNACE, FANCG, FANCI, FNACJ, FANCL, FANCM, FANCN.
שמונה מתוך החלבונים הללו, המקודדים על ידי הגנים FANCA, FANCB, FANCC, FANCE, FANCEF, FANCG, FANCL, FNACM, יוצרים קומפלקס גרעיני הקרוי FA core complex (תמונה 1). היווצרות קומפלקס זה נחוצה לשפעול של FANCD2 לצורתו ה-monoubiqutinated (FANCD2-Long = FANCD2-L).
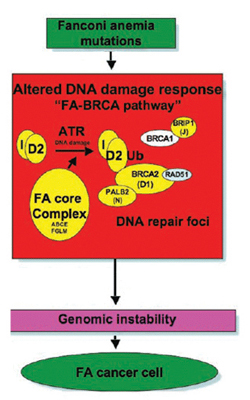
תמונה 1. חלבוני פ"א והנטייה לממאירות הומוזיגוטיות למוטציות בגנים המעורבים בא"פ גורמת להפרעות במסלול תיקון נזקים בדנ"א (FA/BRCA). הקומפלקס הפרוקסימלי (A, B, C, E, F, G, L, M) נחוץ ליובקוטינזציה של ו I-D2. עם ההפעלה של I-D2-Ub החלבונים הללו מכוונים לאזורי תיקון דנ"א בתא(DNA repair foc'). שם הם מתקשרים עם חלבונים אחרים המשתתפים בתיקון דנ"א (BRCA2, RAD51). כאשר נפגע מנגנון תיקון נזקים בדנ"א, יש יציבות נמוכה של הגנום והופעה של ממאירויות
FANCD2-L מתקשר לחלבונים תאיים המשתתפים בתיקון נזקים בדנ"א, כולל BRCA2 ו-.RAD51 לאחרונה התברר ש-FANCI כמו FANCD2 קושר יוביקוויטין. חולי א"פ מקבוצת FA-D1 נושאים שתי מוטציות ב-.BRCA2 כל הממצאים הללו מצביעים על כך שחלבוני פנקוני קשורים למסלול תיקון נזקים בדנ"א הקרוי עתה מסלול FA/BRCA. הפרעה במסלול של FA/BRCA מופיעה בכל אחד מחולי א"פ עקב ירידה בפעילות של אחד מ-13 החלבונים הללו ועקב כך תגובה לא תקינה לנזק לדנ"א ונטייה לפתח גידולים ממאירים. למרות שרבים מהשלבים של המסלול FA/BRCA ידועים, התפקיד המדויק של חלבוני א"פ בתיקון נזקים בדנ"א עדיין לא ברור. יש לציין גם שלתאים מחולי א"פ יש הפרעות נוספות כרגישות לחמצן וקינטיקה לא תקינה של מחזור התא. הסיבה לפגמים אלה עדיין לא הובררה.
התמונה קלינית
א"פ מאופיינת בשכיחות גבוהה של מומים מולדים, בהופעה בעשור הראשון לחיים של כשלון מח עצם שהולך ומתקדם וכן בהתפתחות ממאירויות המטופואטיות בעשור השני וגידולים מוצקים בעשור השלישי לחיים.
מומים מולדים - לרוב החולים עיכוב בגדילה. העיכוב בגדילה מתחיל כבר בחיים התוך רחמיים ורבים מהם נולדים במשקל קטן לגיל ההריון. ממצאים שכיחים נוספים כוללים עיניים קטנות והיקף ראש קטן. לרבים מהחולים אזורי היפו-והיפרפגמנטציה על פני העור. לחולים גם מומים בשלד, האופייניים ביותר הם חסר אגודל או חסר רדיוס ואגודל. תוארה גם שכיחות גבוהה של מומים כלייתיים ככליית פרסה וכליה אגנית וכן מומים בדרכי העיכול כדוגמת אטרזיה של התריסריון. יש לציין של-30 אחוז מהחולים אין מומים כלל.
כשלון מתקדם של מח העצם - תחילתו של כשלון מח העצם בעשור הראשון לחיים, בדרך כלל עם הופעת טרומבוציטופניה קלה שהולכת ומחמירה, מלווה בהמשך בלויקופניה, נוטרופניה ואנמיה. עוד לפני התפתחות טרומבוציטופניה משמעותית ניתן לראות ברבים מהחולים הגדלה לא מוסברת בנפח הכדורית האדומה (macrocytosis) ו/או עלייה בהמוגלובין F. נמצא שעד גיל 40 שנה, הרוב המכריע של החולים (90 אחוז) עם א"פ יפתחו כשלון מח עצם. נמצא גם שמומים מולדים רבים מראים על דרגת מחלה קשה יותר, מלווה בהופעה מוקדמת יותר של כשלון מח עצם.
ממאירות המטולוגית וגידולים מוצקים - בעשור השני לחיים נוטים החולים לפתח ממאירות של מוח העצם ובעיקר לוקמיה מילובלסטית חריפה. שיעור החולים שיפתח ממאירות המטופואטית אינו ידוע, אולם מוערך כי ההיארעות המצטברת של ממאירות המטולוגית עד גיל 40 היא 33 אחוז. הטיפול בממאירות כאשר התגלתה הוא קשה ביותר בגלל חוסר הסבילות של החולים לכימותרפיה. כדי לגלות שינויים טרום ממאירים, על החולים לעבור בדיקת מח עצם ובדיקת כרומוזומים במח העצם פעם בשנה.
הגידולים המוצקים השכיחים הם מסוג שאת של תאי קשקש. האתרים הנפוצים הם ראש-צוואר, דרכי העיכול ואיברי המין בנשים ]כעריה (vulva) וצוואר הרחם[. לפי ניתוח שעלה ממחקר עוקבה רטרוספקטיבי שבחן 144 חולים עם א"פ, קיים במחלה סיכון מוגבר פי 785 מהאוכלוסיה הכללית לפתח לוקמיה, פי 700 לפתח סרטן של אזור ראש-צוואר, פי 2,360 לפתח סרטן של הוושט ופי 4,300 סיכון לפתח סרטן של העריה. יצוין כי בכ-25 אחוז מהמקרים, אבחנה של א"פ נעשית רק לאחר שאובחנה ממאירות. בשל חוסר הסבילות לכימותרפיה, הטיפול בחולי א"פ שפיתחו גידולים הוא קשה והפרוגנוזה רעה. על מנת לגלות מוקדם ככל האפשר את הגידולים הממאירים, בכדי שניתן יהיה לטפל בכירורגיה בלבד, על החולים מגיל 15 והלאה לעבור בדיקות סריקה תקופתיות בחיפוש מתמיד אחרי גידולים אופייניים כולל לרינגוסקופיה לא ישירה, אזופגוסקופיה ובדיקה גניקולוגית בנשים, בתדירות של כפעם בחצי שנה.
הקשר בין מעורבות הגנים השונים במחלה לחומרתה
כ-70-65 אחוז ממקרי א"פ נגרמים כתוצאה מפגם בגן, FANCA לעשרה אחוזים מהחולים יש מוטציה בגן FANCC ולכשמונה אחוזים - בגן- FANCG. מעורבות שאר הגנים נדירה. קבוצת השלמה אחת (FA-B) מורשת בתאחיזה לכרומוזום-X. בגן FANCA תוארו עד כה יותר מ-100 פגמים הגורמים למחלה, רבים מהם חסרים גדולים בגן. מוטציה אחת ב-(IVS4+4 A >C) FANCC (מופיעה ביותר מ-80 אחוז מהיהודים האשכנזים. שכיחות הנשאים למוטציה זו בקרב יהודים אשכנזים היא 1.1 אחוז.
מתקבל הרושם שהתמונה הקלינית של חולים מקבוצת השלמה C ו-G חמורה יחסית בהשוואה ל-קבוצת A, הן מבחינת מספר המומים והן מבחינת חומרת כשלון מח העצם. חולים עם פגיעה ב- BRCA2) FANCD1 ) סובלים מתמונה קשה יותר עם הופעת ממאירויות בשנים הראשונות לחיים, כולל לוקמיה מילובלסטית חריפה, גידולי מוח וגידולי כליה.
אבחנה
בכל הילדים עם מומים מולדים אופייניים או ממצאים המטולוגיים מחשידים (מאקרוציטוזיס, טרומובציטופניה או פנציטופניה) ובוודאי בנוכחות שניהם, יש לחשוד בא"פ ולבצע את המבחן האבחנתי, מבחן להשראת שבירות כרומוזומים. קביעת הגן הפגום היא משימה סבוכה יותר, כפי שיוסבר להלן.
מבחן להשראת שבירות כרומוזומלית (chromosomal breakage test) - מבחן זה הכרחי לאבחנת א"פ. במבחן מדגירים תאים שמתחלקים במהירות, לרוב לימפוציטים מדם פריפרי שעוררו על ידי פיטוהמאגלוטינין
(PHA) או על ידי פיברובלסטים מהעור, בנוכחות מינונים נמוכים של mitomycin C) MMC) או diepoxybutane (DEB). לאחר מכן בוחנים את הכרומוזומים במיטוזה ומחפשים שברים. בחולי א"פ יש מספר שברים מוגבר גם ללא חשיפה לחומרים קושרי גדילי דנ"א, אך הוא עולה משמעותית לאחר תוספת החומרים הללו. אם המבחן שלילי כאשר מבוצע בלימפוציטים ובכל זאת החשד הקליני גבוה, יש לבצעו גם בפיברובלסטים. מוכרים מצבים של מוזאיקה שבהם מוטציה סומטית תיקנה את הפגם בתא גזע המטופואטי.
Immunoblotting לנוכחות החלבון FANCD2-L - בדיקה שבה נקבעת נוכחות החלבון. הבדיקה מאפשרת להבדיל האם הגן הפגוע הוא פרוקסמלי או דסטלי לקשירת היוביקויטין ל-FANCD2. העדר FANCD2-L מכוון למוטציה באחד מהגנים המקודדים לשמונת חלבוני הקומפלקס
(A, B, C, E, F, G, L, M). הימצאות FANCD2-L מעידה על פגם בגן המקודד לחלבונים הדסטליים J ,(BRCA2) D1 או N.
זיהוי המוטציות - זיהוי המוטציות בכל חולה הוא התנאי לאפשרות לבצע אבחנה טרום לידתית ויכול לעזור לצפות את המהלך הקליני ולכוון את מועד השתלת תאי האב ההמטופואטיים. מומלץ לכן לנסות ולאתר את הגן הפגוע בכל חולה.
Ameziane וחבריו הציעו את הגישה הבאה לזיהוי מוטציות בחולי א"פ. לאור השכיחות היחסית של מוטציות ב-FANCA להתחיל עם סקירתו של גן זה, הם השתמשו בשיטת MLPA Multiplex Ligation-dependent Probe)Amplification), שיטה מסחרית MCR (Holland BV, Amsterdam, The Netherlands) המאפשרת לקבוע את מספר המקטעים המוכפלים וכך לזהות חסרים בגן FANCA. במקביל לחיפוש חסרים גדולים בגן, הם הציעו לבצע ריצוף של האזורים המקודדים בגן FANCA (על 43 האקסונים שלו).
במקרה של חולים זכרים בלבד במשפחה, יש לחשוד ולסרוק בשלב ראשון את FANCB. בחולים בהם התורשה אוטוזומלית רצסיבית, לאחר שלילית פגמים בגן FANCA, מומלץ לסרוק את FANCC, FANCE ו-FANCF.
אם FANCD2-L קיים, יש להתרכז בגנים BRCA2, FANCN ו-FANCJ. גישתנו לבירור הבסיס הגנטי של חולי א"פ שונה במקצת, בהתבסס על פגמים אופייניים שמצאנו בקבוצות אתניות שונות בארץ, כפי שיוסבר להלן בפרק על א"פ בישראל.
מניעת המחלה - אבחנה טרום לידתית
היות שמדובר במחלה קשה ביותר, יש לעשות הכל כדי למנוע היוולדות ילדים חולים נוספים במשפחות שבהן יש כבר ילד חולה. יש לעשות מאמץ לזהות את המוטציה ומשזו מזוהה, ניתן לבצע בדיקת סיסי בשליש הראשון להריון (שבוע 12-10), להפיק דנ"א ולקבוע האם העובר נגוע. במקרה של עובר נגוע, יש להמליץ על הפסקת הריון מוקדמת.
Preimplantation Genetic Diagnosis) PGD) מאפשרת בדיקת העובר לפני השרשה בכדי להבטיח שאינו נושא את המחלה וכן אפשר לבדוק האם הוא מתאים מבחינת סיווג רקמות לחולה, כך שיוכל לשמש בעתיד תורם לתאי אב המטופואטיים.
טיפול
הסיבה העיקרית לתמותה בא"פ היא כשלון מח עצם. אנדרוגנים אנבוליים יכולים לגרום לעלייה בספירות הדם ב-50 אחוז עד 70 אחוז מהחולים, אך רובם יפתחו עמידות לטיפול זה. השתלת תאי אב המטופויטיים מתורם מתאים מבחינת סיווג הרקמות היא הטיפול היחידי המאפשר ריפוי של כשלון מח העצם והורדת סיכון להתפתחות ממאירות המטולוגית.
השתלת תאי אב המטופויטיים - תאי חולי א"פ רגישים לטיפול כימותרפי כמו ציטוקסן וקרינה. פרוטוקולים ראשוניים של הכנה שכללו ציטוקסן וקרינה לוו בטוקסיות רבה ובתוצאות גרועות. רק ירידה במינון ציטוקסן לעשירית ממינון הניתן בילדים ללא א"פ (20 מ"ג לק"ג) הביאה לשיפור התוצאות בהשתלת תאי אב המטופויטיים. שיעור החיות לאחר חמש שנים בקרב חולי א"פ שהושתלו מתורם המתאים מבחינת סווג רקמות הוא 85 אחוז. התוצאות לגבי תורם מתאים שאינו בן משפחה טובות הרבה פחות ועומדות על 20 אחוז עד 40 אחוז לאחר שנתיים. כדי לשפר את התוצאות, נעשים שינויים בפרוטוקול ההכנה. פרוטוקולים המכילים פלודארבין, ללא קרינה או עם כמות קטנה של קרינה, מראים תוצאות ראשוניות מעודדות.
יש לציין עם זאת שמעקב ארוך טווח אחרי חולים שעברו השתלת תאי אב המטופויטיים מראה שכיחות גבוהה של ממאירות במיוחד של ראש-צוואר, שמונה עד עשר שנים לאחר השתלה. גידולים אלה מופיעים מוקדם יותר בחולים שעברו השתלה יחסית לחולים שלא עברו השתלה. תופעה קשה זו קשורה ככל הנראה לנטייה של חולי א"פ לממאירות כחלק מאי יציבות הגנום, ומוחמרת על ידי השימוש ברדיותרפיה.
הישרדות - ההישרדות החציונית של חולים עם א"פ בסדרות שונות נעה בין 24 שנה ל-30 שנה. גיל הופעת הסיבוכים משתנה ותלוי, בין השאר, במין החולה ובסוג המוטציה. לאור אפשרויות הטיפול באי ספיקת מח עצם ומניעת הממאירות ההמטולוגית בעזרת השתלת תאי אב המטופויטיים, הסיבוך העיקרי כיום הוא הופעת גידולים ובעיקר גידולים מוצקים.
אנמיה על שם פנקוני בישראל
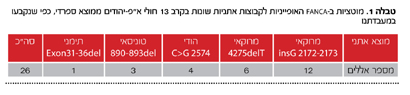
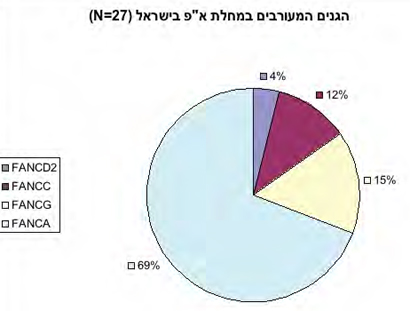
במהלך השנים נלמדו במעבדתנו הפגמים המולקולריים האחראים לא"פ בישראל. תיארנו ארבע מוטציות ב-FANCA, הקשורות במוצא האתני ונמצאו אצל יהודים ממוצא ספרדי (מרוקאים, תוניסאים והודים). סך הכל נבדקו במעבדתנו בעשר השנים האחרונות 40 חולים, ש-27 מהם אינם קרובי משפחה. פיזור הגנים המוטנטים בקבוצת חולים זו מופיע בתמונה 2. במרבית החולים נמצאו מוטציות בגן 69) A אחוז), ב-15 אחוז מהמוטציות בגן G וב-12 אחוז בגן C. רק בחולה אחד נמצא פגם בגן FANCD2. מתוך 27 החולים, 17 היו יהודים ועשרה ערבים. מתוך 17 היהודים, ארבעה ממוצא אשכנזי, שלושה עם המוטציה האשכנזית ואחד עם שינוי ב-BRCA2. הפיזור של הפגמים ב-13 החולים היהודים הנותרים ממוצא ספרדי התאים לקבוצה האתנית שלהם, כמוצג בטבלה 1. אשר לעשרת החולים ממוצא ערבי, שישה נשאו מוטציות FANCA (שני חסרים גדולים וארבע מוטציות נקודתיות) וארבעה פגמים ב-FANCG (כולם חסרים נקודתיים). כל הפגמים הללו, להוציא אחד, הם ייחודים ולא תוארו בעבר בחולי א"פ.
בהתבסס על מידע זה, גישתנו לבירור מקרה חדש היא שבחולים ממוצא יהודי אתני מוגדר ניתן לקבוע ישירות את קיום המוטציה האופיינית (לאשכנזים, יהודים ממוצא מרוקאי וכו'). בחולים שאינם נמנים על אחת מהקבוצות האתניות ויש להם פגמים ייחודיים, יש לבצע קביעה לקיום של החלבון FANC2-L. אם FANCD2-L חסר, מדובר באחד משמונת הגנים של הקומפלקס הפרוקסמלי (תמונה 1). היות שרוב המוטציות בעולם וגם בישראל הן בגן A, בשלב זה אנו בודקים את הגן FANCA, זאת על ידי קביעת חסרים גדולים בשיטת MLPA, ואם אלה לא קיימים, קביעת רצף הבסיסים בגן על 43 האקסונים שלו. אם לא נמצאה מוטציה בגן FANCA, המועמד הבא הוא הגן FANCG. כאשר שבירות הכרומוזומים מוגברת ו-FANCD2-L קיים, הפגם קרוב לוודאי מצוי באחד הגנים הדיסטלייםBRCA2 FANCN) FANCD1 או FANCJ ).
תמונה 2: פיזור הגנים שבהם נמצאו מוטציות בחולי א"פ שנבדקו במעבדתנו. מתוך 27 חולים שנבדקו במעבדתנו ושאינם קרובי משפחה, ל-69 אחוז היתה מוטציה ב-FANCA, ל-15 אחוז ב-FANCG ול-12 אחוז ב-FANCC
לסיכום, בשנים האחרונות חלה התקדמות רבה בהבנה של א"פ ובחשיבותם של חלבוני פנקוני בשמירת יציבות הגנום. התברר שא"פ נגרמת על ידי מוטציה באחד מלפחות 13 גנים שונים. המחלה מאופיינת בריבוי מומים מולדים ובנטייה להתפתחות כשלון מח עצם ולהופעת גידולים ממאירים הן במערכת ההמטופויטית והן גידולים מוצקים. כשלון מח העצם והנטייה לממאירות המטולוגית ניתנים לטיפול בחלק מהמקרים בעזרת השתלת תאי אב המטופויטיים. בחלק מהחולים, לאחר השתלות כאלו יש נטייה להופעת מוקדמת של גידולים ממאירים. על החולים בסיכון לעבור סריקות תכופות לגילוי הגידולים האופייניים. יש לעשות כל מאמץ לגלות את הגן ואת המוטציה הגורמת למחלה בכל חולה, על מנת לחזות את חומרת המחלה וגם למנוע היוולדות ילדים חולים נוספים במשפחה.
ד"ר דניאלה נשרי, מחלקת ילדים ג', פרופ' חנה תמרי, היחידה להמטולוגיה, מרכז שניידר לרפואת ילדים בישראל |